A research team led by Chair Professor Chuxia Deng and Assistant Professor Miao Kai in the Faculty of Health Sciences (FHS) at the University of Macau (UM) has made a significant breakthrough in the field of cancer immunotherapy. The study unveils for the first time how the key gene, dual-specificity phosphatase 22 (DUSP22), enhances T cell infiltration into tumours by ‘trimming the stability tag’ of the immunosuppressive factor LGALS1. This discovery not only provides a novel perspective on tumour immune evasion but also proposes a promising combination strategy to overcome immunotherapy resistance in ‘cold’ tumours, such as triple-negative breast cancer (TNBC). The findings have been published in the Journal for ImmunoTherapy of Cancer, the official journal of the Society for Immunotherapy of Cancer (SITC).
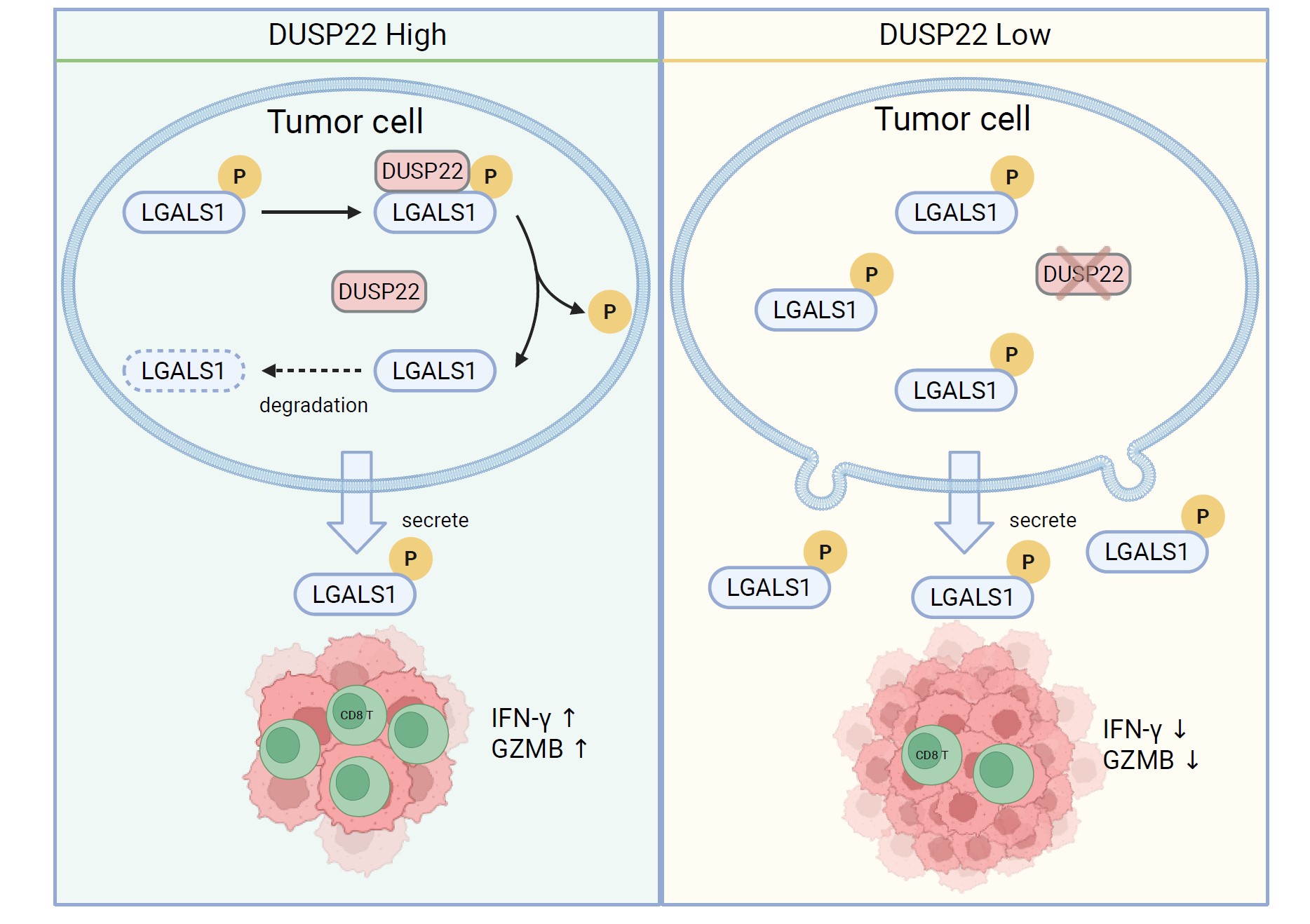
In the tumour microenvironment, the scarcity of CD8⁺ T cells—a primary type of anti-cancer immune cell—is a fundamental reason for the poor efficacy of immunotherapies against many solid tumours. Transforming these immune-desert ‘cold’ tumours into treatment-sensitive ‘hot’ tumours is a key challenge in cancer immunotherapy today. To address this, the team used genome-wide screening techniques to identify 39 candidate genes associated with T cell infiltration in a mouse model of breast cancer. Among these, DUSP22 emerged as the most prominent gene. Experiments confirmed that the level of DUSP22 in breast cancer tissues positively correlates with CD8⁺ T cell infiltration and negatively correlates with malignant tumour progression. Crucially, overexpressing DUSP22 in tumour cells significantly suppressed tumour growth and prolonged the survival of mice with a competent immune system, an effect that was completely absent in immunodeficient mice. This is akin to discovering a ‘switch’ within tumour cells that regulates immune cell infiltration into solid tumours. The team also elucidated the mechanism by which DUSP22 specifically removes phosphate groups from two key sites (Ser8/Thr58) on the immunosuppressive molecule LGALS1. This dephosphorylation destabilises LGALS1, leading to its rapid degradation. As LGALS1 typically induces T cell apoptosis and hinders their ability to cross blood vessels and enter tumours, its depletion relieves immunosuppression, allowing T cells to infiltrate tumours unimpeded and kill cancer cells efficiently.
Based on this mechanism, the team further explored its clinical translational potential. They found that LGALS1 inhibitors (such as OTX008) and neutralising antibodies not only inhibit tumour growth independently, but also demonstrate a significant synergistic effect when combined with the widely used anti-PD-1 immunotherapy. This combination therapy successfully transformed immunotherapy-resistant ‘cold’ tumours into responsive ‘hot’ tumours, offering a new avenue to overcome immunotherapy resistance. This research expands the function of DUSP22 beyond traditional signal transduction regulation, establishing its crucial role in the ‘crosstalk’ between tumours and the immune system. It also highlights the critical role of LGALS1 phosphorylation in tumour immune evasion.
The corresponding authors of this study are Prof Deng and Prof Miao. The first author is Wang Lijian, a PhD student in UM FHS. The research team also includes scholars from UM, the University of Hong Kong–Shenzhen Hospital, the Macau University of Science and Technology, and the Third Affiliated Hospital of Zhengzhou University. The project was supported by the Science and Technology Development Fund of the Macao SAR (File Nos.: 0073/2021/A2, 111/2017/A, 0007/2021/AKP, 0087/2024/RIB2, 0009/2022/AKP, 0065/2021/A, 0193/2024/AGJ), the University of Macau (File Nos.: MYRG-GRG2023-00150-FHS-UMDF, MYRG-GRG2024-00146-FHS), and the National Natural Science Foundation of China (File No.: 82030094). The full article is available at: https://doi.org/10.1136/jitc-2025-013142
| Source: Faculty of Health Sciences | |
| Media Contact Information: | |
| Communications Office, University of Macau | |
| Albee Lei | Tel: (853) 8822 8004 |
| Bell Leong | Tel: (853) 8822 8009 |
| Email: | prs.media@um.edu.mo |